How Residue Substitutions Modulate Metabolism Speed in Human NAT2 Enzyme: Insights from Molecular Dynamics Simulations
Dr Wayde Veldman
Abstract Authors
Wayde Veldman - Research Unit in Bioinformatics (RUBi), Department of Biochemistry, Microbiology and Bioinformatics, Rhodes University
Ozlem Tastan Bishop - Research Unit in Bioinformatics (RUBi), Department of Biochemistry, Microbiology and Bioinformatics, Rhodes University
Abstract Description
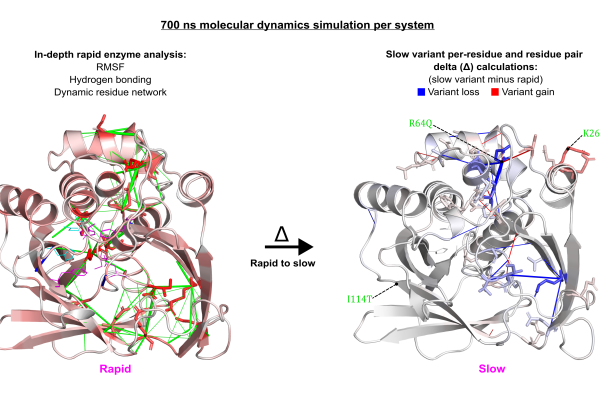
Investigating how residue substitutions impact the structure, interactions, and dynamics of drug-metabolising enzymes remains an underexplored area, yet it holds great potential for understanding challenges related to drug resistance, efficacy, and toxicity. In this study, we performed all-atom molecular dynamics simulations to uncover detailed mechanistic insights of arylamine N-acetyltransferase 2 (NAT2), the enzyme responsible for metabolising the TB drug isoniazid. Our aim was to understand how residue substitutions drive the transition from rapid to slow metabolism. We developed a computational framework to compare rapid and slow enzymes using analyses of hydrogen bonding, residue fluctuation, and dynamic residue network centrality. In general, slow-metabolising variants showed destabilisation of the active conformation, driven by altered hydrogen bonding and disrupted communication within structural networks, resulting in increased local flexibility. For example, in the R64Q+K268R variant, the R64Q residue substitution led to a significant loss of hydrogen bonds, leading to increased residue flexibility and disrupted network centrality. Key functional residues—including catalytic D122 and putative isoniazid-binding residues S125 and F217—exhibited significantly reduced betweenness centrality. On the other hand, eigenvector centrality decreased for residues N72, D122, and active site loop G124. These decreases of network centrality indicate a communication loss in the active site. Furthermore, this variant showed a substantial shortening of the average distance between binding residue F217 and catalytic residues H107 and D122, indicating a shift in active site geometry. Altogether, our results offer mechanistic insight into how allosteric residue changes impair enzyme function and ultimately modulate drug exposure.Dr Wayde Veldman
RUBi
