Abstract Authors
Bridget Valeria Zinhle Nkosi & Özlem Tastan Bishop
Research Unit in Bioinformatics (RUBi), Department of Biochemistry, Microbiology and Bioinformatics, Rhodes University, South Africa
Abstract Description
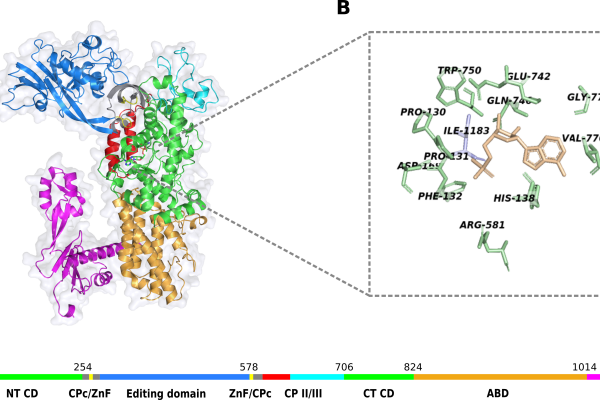
Malaria remains a major global health challenge, compounded by increasing drug resistance and the toxicity of existing therapies. This study addresses these challenges by targeting cytoplasmic Plasmodium falciparum isoleucyl-tRNA synthetase (cPfIleRS), an essential and highly conserved enzyme previously identified as a potential drug target. We investigated a class of thienopyrimidines, previously reported by Isvan et al. (PMCID: PMC10286833) as multi-stage inhibitors of Plasmodium vivax IleRS. Ten thienopyrimidine compounds were initially screened for inhibitory activity against cPfIleRS. From these three promising candidates, MMV007938, MMV019904, and MMV019837 (Nkosi et al., unpublished) were identified and subsequently referred to as Parent 1, 2, and 3, respectively. These were used to generate analogue libraries via an in-house RDKit script. Through structure-based computational methods and calculations, including molecular docking, molecular dynamics simulations, RMSD, comparative essential dynamics, centre of mass, RMSF, and MM/GBSA binding free energy calculations. We identified four analogues (1PAN29, 1PAN31, 1PAN44, and 3PAN8) with strong binding affinities and evident effects on cPfIleRS structure and function. To tackle drug toxicity, a selectivity analysis against the human homologue (HsIleRS) was performed. This revealed a preferential inhibitory effect on cPfIleRS, suggesting favourable selectivity. Interestingly, these compounds all bound to potential allosteric sites. They induced increased residue fluctuations, particularly in the editing domain, agreeing with potential allosteric inhibition. Remarkably, perturbations in the AMP and isoleucine-binding pocket (active site) disrupted critical residue interactions, and, in some cases, the isoleucine substrate was removed from the active site. MM/GBSA analysis further confirmed reduced AMP binding affinity upon ligand binding. Per-residue MM/GBSA decomposition analysis identified key residues essential for cPfIleRS function, including HIE138, GLY140, HID141, TRP214, ARG581, GLU742, and the KMSK motif (LYS783, LYS786). Compound binding interfered with residues interacting with AMP, suggesting potential allosteric interference with aminoacylation. Others induced structural changes and misalignment of the catalytic residues. These results support the role of these compounds as allosteric modulators capable of disrupting cPfIleRS function. These findings support the potential of thiophene-fused pyrimidine analogues as selective antimalarial agents targeting cPfIleRS, providing a promising foundation for future drug development.

